- Research
- Open access
- Published:
Transcriptional profiling of predator-induced phenotypic plasticity in Daphnia pulex
Frontiers in Zoology volume 12, Article number: 18 (2015)
Abstract
Background
Predator-induced defences are a prominent example of phenotypic plasticity found from single-celled organisms to vertebrates. The water flea Daphnia pulex is a very convenient ecological genomic model for studying predator-induced defences as it exhibits substantial morphological changes under predation risk. Most importantly, however, genetically identical clones can be transcriptionally profiled under both control and predation risk conditions and be compared due to the availability of the sequenced reference genome. Earlier gene expression analyses of candidate genes as well as a tiled genomic microarray expression experiment have provided insights into some genes involved in predator-induced phenotypic plasticity. Here we performed the first RNA-Seq analysis to identify genes that were differentially expressed in defended vs. undefended D. pulex specimens in order to explore the genetic mechanisms underlying predator-induced defences at a qualitatively novel level.
Results
We report 230 differentially expressed genes (158 up- and 72 down-regulated) identified in at least two of three different assembly approaches. Several of the differentially regulated genes belong to families of paralogous genes. The most prominent classes amongst the up-regulated genes include cuticle genes, zinc-metalloproteinases and vitellogenin genes. Furthermore, several genes from this group code for proteins recruited in chromatin-reorganization or regulation of the cell cycle (cyclins). Down-regulated gene classes include C-type lectins, proteins involved in lipogenesis, and other families, some of which encode proteins with no known molecular function.
Conclusions
The RNA-Seq transcriptome data presented in this study provide important insights into gene regulatory patterns underlying predator-induced defences. In particular, we characterized different effector genes and gene families found to be regulated in Daphnia in response to the presence of an invertebrate predator. These effector genes are mostly in agreement with expectations based on observed phenotypic changes including morphological alterations, i.e., expression of proteins involved in formation of protective structures and in cuticle strengthening, as well as proteins required for resource re-allocation. Our findings identify key genetic pathways associated with anti-predator defences.
Background
The common freshwater micro-crustacean Daphnia has become a model organism for many biological disciplines [1–6]. The extensive knowledge of its ecology [5, 7, 8] and its biological responses to environmental changes [3, 9, 10] together with the availability of genomic resources [4] make the system highly attractive for evolutionary ecology research and provides the unique opportunity to study ecological traits with the aid of emerging molecular biological tools. One of the most intriguing ecological responses of Daphnia species to environmental changes is their ability to develop different phenotypes given the same genetic background, a phenomenon called phenotypic plasticity. Prominent examples of phenotypic plasticity include inducible defences.
Inducible defences are interpreted as adaptations to heterogeneous predation risks and are found in many organisms from protists to vertebrates [11–13]. Daphnia evolved sensitivity against specific chemical compounds, which are unintentionally emitted by their predators. These so-called kairomones serve as signals which prompt the daphnid prey to develop individuals which are better defended. Previous work has shown that different predators, e.g. fish and the phantom midge Chaoborus spp., can induce different, sometimes opposite phenotypic reactions in the same species or clone [10, 14–17]. This means that the genome must encode multiple developmental programs triggered by environmental conditions. Induced defences in Daphnia include prominent morphological modifications: from tiny cuticular teeth to very elongated tail and head spines, helmets or even giant crests [18–21], but also changes in life history and different behaviours, which ultimately all act as deterrents to encounter, capture and ingestion by the predator [9, 22–25].
In the model species Daphnia pulex, kairomones from the phantom-midge larvae Chaoborus trigger production of neck-teeth, the most easily detectable trait, and overall hardening of the cuticle [26]. These external, cuticle-associated alterations effectively protect juveniles from predation [27, 28]. At the same time, induced D. pulex females shift resources from reproduction to somatic growth, thereby reaching maturity at a larger size and producing less but larger offspring [29–31]. Vertical migration is deemed to comprise the main behavioural reaction to the presence of the predator in D. pulex: thus, Chaoborus-induced specimens prefer shallower depths in comparison to control specimens [32, 33]. Chaoborus is an ambush predator, such that Daphnia is also expected to reduce its swimming speed, although in the case of D. pulex this habit is displayed only by some clones [34–36] (LCW, unpublished observations).
Instability of environmental conditions (periodicity of predation risk, different predators) and costs of defences explain the inducible nature of the defensive morphs. This is also in line with the fact that the neck-teeth are present only in certain juvenile instars when the daphnids reach preferred prey size of their gape-limited predators [10, 13, 29, 37, 38].
Based on the experimental evidence we can make the following predictions regarding the underlying functional classes of effector genes that might contribute to Daphnia’s anti-predatory response (Fig. 1):
-
1.
the structural changes in the cuticle are expected to mirror changes in the amounts/types of cuticle-associated proteins;
Fig. 1 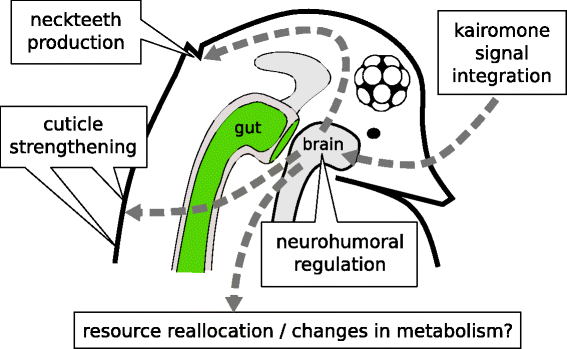
Presumptive scheme of physiological and morphological changes during kairomone-induction in D. pulex
-
2.
life history modifications are expected to be controlled by several physiological changes affecting both somatic growth and reproduction;
-
3.
one can predict that other metabolic functions should be down regulated under predation risk in order to allocate energy resources primarily to the above mentioned pathways;
-
4.
all levels of the response must ultimately be controlled by cascades of receptors, humoral factors, signalling pathways and transcription factors.

Currently, the technique of choice suitable for addressing response patterns in gene expression at a genome-wide level with potentially unlimited depth of coverage is RNA-Seq [39–41]. The availability of the D. pulex draft genome [4] greatly facilitates the power of such analyses in that RNA-Seq reads can be specifically mapped to a particular genomic location. Investigations of the genome-environment interactions in Daphnia are ongoing, with the results of the first analyses of differential gene expression patterns in ecological experiments recently becoming available [42–45]. A number of features have been discovered that point to an ecological responsiveness of the D. pulex genome; e.g. a large overall number of genes, organized in the many families of paralogous genes that in many cases do not show homologies to genes in other organisms, but show differential expression under different environmental conditions [4, 46–49].
While preliminary analysis of the transcriptomic responses of D. pulex to the predator was performed earlier [4], gene expression was assessed with tiling microarrays and was restricted to the second juvenile instar, after the onset of neck-teeth production. Here we aimed at providing the first whole-genome analysis of gene expression changes involved in formation of predator-induced defences in D. pulex. To accomplish this, we apply the most versatile technique to study transcriptomes, RNA-Seq and focus on the first juvenile instar, the developmental point at which the defence is expected to unfold.
Material and methods
Daphnia clone used in the experiment
In contrast to the clone chosen for genome sequencing by the Daphnia Genomics Consortium (“TCO”), the clone we utilized for the experiment (designated as “R9”) is known to show pronounced production of defensive morphs in the presence of the phantom midge larvae Chaoborus [4]. It originates from Canada and according to mitochondrial markers belongs to the so-called Panarctic Daphnia pulex clade. The TCO clone in turn belongs to a group of populations united under the name “Daphnia arenaria” which is likely of hybrid origin with its mitochondrial genome coming from the same clade as R9, while nuclear markers point to closer relationships with North American Daphnia pulicaria [4, 50, 51].
Experiment
We utilized a simple experimental design: one pooled series of Daphnia pulex juveniles exposed to Chaoborus and a control set of specimens without predator induction. Fifty age-synchronized specimens of the D. pulex R9 clone, served as the founding generation for the experimental animals. For induction, the mothers bearing late embryos were exposed to the Chaoborus larvae contained in a net cage and fed with 100 juvenile daphnids. Progeny obtained from the induced and control mother specimens was collected at the first juvenile instar, and stored in RNA Later (Qiagen, Hilden, Germany) for 24 h. Subsequently, specimens were stored at -20 °C until RNA extraction. Three independent induction experiments were performed leading to 90 juveniles in each group (induction and control) in total, which were pooled together to level individual variation.
RNA extraction and sequencing
Total RNA was extracted with the TRIzol Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol with modifications. DNA was further depleted with DNase treatment (TurboDNase, Life Technologies, Carlsbad, CA, USA) and its absence was confirmed afterwards via PCR with primers spanning exon-intron boundaries in an α-tubulin gene. Quality and amount of the purified RNA in the samples was analyzed on the Experion System (Bio-Rad, Hercules, CA, USA) with the aid of the Experion RNA StdSens Analysis Kit according to the supplied manual. The samples were shipped to Otogenetics (Norcross, GA, USA) for library preparation and sequencing. cDNA was synthesized with random hexamers after rRNA depletion. Size-selected cDNA fragments (250–300 bp range) were sequenced on an Illumina HiSeq 2000 from both ends. Overall, two sequencing runs were performed yielding 10–20 million 100 bp read pairs per sample.
Quality of the reads was analyzed with FastQC v. 0.10.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and necessary filtering steps were performed with trimmomatic v. 0.22 (http://www.usadellab.org/cms/index.php?page=trimmomatic). Since a considerable contamination of the data with adapter sequences and non-coding RNAs (primarily, rRNA) was detected in the first experiment, we performed two rounds of trimmomatic treatment: removal of reads showing similarity to sequences of non-coding RNA (100 bp tiling fragments with 50 bp overlaps were used as queries) (ILLUMINACLIP:non_coding_rnas:4:40:12 MINLEN:81, for positive matches the whole pair was discarded) and adapter and quality trimming on the second step (ILLUMINACLIP:adapters:2:40:9 LEADING:15TRAILING:15MINLEN:36). After each step of contamination removal a quality control analysis using FastQC was performed to check the validity of our data processing steps.
The raw reads are available from the NCBI Sequence Read Archive (BioProject accession number: PRJNA287609).
Assembly
We employed two principal alternative approaches to assemble the transcriptome: mapping-oriented and de novo assembly. For mapping we took the latest set of scaffolds for the Daphnia pulex genome: the 06.09.2005 version with further filtering steps as available on http://genome.jgi-psf.org/Dappu1 (5,191 scaffolds with the total length of 197,261,574 bp). “Daphnia pulex Genes 2010 beta 3” annotations, provided by the wFleaBase (http://wfleabase.org) were utilized for the reference-genes-guided steps. Those loci which were located on the filtered out scaffolds were excluded, and the final gene set comprised 41,561 transcripts in total. Intron length boundaries were estimated based on the official gene annotations.
Mapping of the reads was performed independently with TopHat v. 2.0.6 [52] and GSNAP v. 2012-12-20 [53]. In TopHat the following principal options were specified for mapping paired reads: --read-mismatches 7 --read-gap-length 2 --read-edit-dist 7 --mate-inner-dist 0 --mate-std-dev 100 --min-anchor-length 5 --min-intron-length 10 --max-intron-length 50000 --max-insertion-length 3 --max-deletion-length 3 --microexon-search --segment-mismatches 3 --segment-length 18 --no-coverage-search --min-segment-intron 10 --max-segment-intron 50000 --min-coverage-intron 10 --max-coverage-intron 20000 --b2-sensitive --report-secondary-alignments --max-multihits 10. Singleton reads decoupled during read filtering were mapped independently with analogous parameters, except that no new junctions were allowed (--no-novel-juncs), besides junctions, obtained on the first step, as recommended by the developers. Mapping options for GSNAP were as follows: --max-mismatches=0.07 --suboptimal-levels=2 --novelsplicing=1 --localsplicedist=50000 --pairmax-rna=50000 --sam-multiple-primaries with the paired reads and singletons analyzed together.
Transcripts for both mapping approaches were assembled with Cufflinks v. 2.0.2 [54, 55]. The assembly was performed with the following general parameters: --multi-read-correct --upper-quartile-norm --max-intron-length 50000 --min-intron-length 10 --overlap-radius 10 and the bias correction option turned on. Two alternative assemblies were made: de novo assembly without predefined annotations and reference-guided assembly (the --GTF option) with cuffmerge and cuffcompare, respectively, used to compare the reconstructed transcripts to the reference.
Mapping-independent de novo assembly was done with Trinity r. 2012-10-05 [56]. Input dataset for Trinity was prepared as follows. First, for the paired reads in each pair an attempt was made to merge the mates in one longer fragment with the requirement of at least 15 bases of overlap and allowance of no more than 2 % mismatches, with the fastq-join program from the ea-utils package v. 1.1.2 (http://code.google.com/p/ea-utils). Resulting merged fragments, remaining paired reads and singleton reads were treated as unpaired sequenced and were united together both from the control and treatment with subsequent exclusion of redundant identical sequences by fastx_collapser, a program from the FASTX-Toolkit v. 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit). Resulting dataset was processed by Trinity with defaults settings, with the exception of butterfly --max_diffs_same_path=5 --min_per_align_same_path=0.95 -SW options and minimal contig length of 60 nucleotides. To obtain expression levels for the Trinity contigs, individual reads were aligned back to them with the aid of Bowtie2 v. 2.0.4 [57] with --end-to-end --sensitive --rdg 7,3 --rfg 7,3 settings separately for the control and the treatment. Resulting mappings were processed by cufflinks with the following parameters: --multi-read-correct --upper-quartile-norm --overlap-radius 10 and the whole contigs specified as predefined annotations, as well as input for bias correction. To define genomic positions of the Trinity contigs, we mapped them to the reference genome with blat (default settings for RNA and minimal identity 93 %). To detect presence and classes of overlaps of the Trinity contigs and the official annotations, cuffcompare from the cufflinks package was used.
Differential expression on the level of genes was assessed with cuffdiff with parameters compatible with those of cufflinks.
Functional annotations
Two different sources of information on gene functions were used:
-
Official annotations as supplied with the “Genes 2010 beta 3” gene set.
-
InterProScan [58] motif-based analysis for the whole official gene set was performed with the RunIprScan v. 1.0.0 client (http://michaelrthon.com/runiprscan/) in April 2013.
Accordingly, two sets of gene ontology (GO) assignments were used for GO-term enrichment analysis: from the official gene annotations and from the information provided by InterPro for individual domains and families.
Predictions made with RunIprScan were expanded to all members of the respective families of paralogous genes. These families were defined based on pairwise similarity of the amino-acid sequences of all genes longer than 16 amino-acids with the aid of blat, similar to the approach utilized for detection of potentially indistinguishable cDNA sequences outlined above. A pair of proteins were assigned to the same family if alignment segments with a minimum of 60 % identity covered at least 90 % of the longer protein sequence, which can be considered as a conservative and safe approach (compare with [59]).
Term enrichment analyses for GO-terms, gene families and InterPro domain and family assignments were performed in GOseq v. 1.12.0 [60] for lists of differentially expressed genes obtained with mapping-based approaches only. All genes we weighted by their length and the resulting probability weighting function was used for the over-representation analysis with default settings, independently for up- and down-regulated genes. Only genes with respective annotations (GO classes, gene family membership etc.) were involved in the calculation. Lists of over-represented terms were filtered by controlling false discovery rate at the level of 0.025 within tests.
Assigning biological function to genes
The multitude of genes without identifiable orthologs in other animal genomes makes correct functional characterization of Daphnia genes challenging. In the current work, we overcame this obstacle in two ways. First, we used not only the gene function assignments as reported in the official gene prediction set, but also results from an independent domain-oriented analysis (InterProScan). The second strategy was to expand these functional designations to whole families of paralogous genes defined in a conservative way. This last approach inevitably leads to propagation of false positives, but nevertheless it provides a good starting point for gene function prediction in a situation when no other source of evidence is available. The D. pulex genome contains an extraordinarily large number of genes organized in families of paralogs [4]. Given this multiplicity of gene families, even with sensitive mapping-oriented transcriptome assembly, there is always uncertainty about precise locations of the reads coming from genes which are very similar on the mRNA sequence level. This problem becomes even more evident when dealing with D. pulex clones different from the one chosen for genome sequencing, as was the case in this study. If not compensated, this can lead to incorrect estimations of differential expression as well as biased results with regard to enrichment analyses. As described in detail below, we decided to select only a single gene from a group if the similarity between their transcripts exceeded a given threshold, 90 % of a pairwise alignment covered by 96 % identical segments.
Detection of potentially indistinguishable cDNA sequences
Highly similar cDNA sequences were detected with blat [61]: mRNA sequences from the reduced official gene set were used as the query and the target in the same run. blat alignment segments with 96 % identity for individual hits were merged and pairs with overall alignment length at least 90 % were detected as potentially indistinguishable. Individual groups of highly similar genes were created based on the resulting network of pairwise hits. For down-stream analyses only single representatives of the respective groups were utilized.
Analysis backbone
All of the necessary format conversions and file rearrangements were performed with the aid of SAMtools v. 0.1.18 [62], standard Unix commands, MySQL queries and custom PHP, R and bash scripts. The data and the reference genome were visualized and manipulated on a local installation of the UCSC Genome Browser [63] with the assembly 06.09.2005 of the Daphnia pulex genome [4] and associated annotations (raw files were downloaded from the wFleaBase: http://wfleabase.org). MySQL-database for the genome browser was further customized to incorporate the results of our functional annotations and information on the lists of differentially expressed genes (see above) and besides the standard interface was accessed directly with SQL commands and a local web-based search tool. Venn and pie diagrams were constructed with the aid of the VennEuler v. 1.1-0 R package [64] and the SVGGraph library (http://www.goat1000.com/svggraph.php), respectively.
Results
RNA-Seq data quality
After the stringent filtering steps, 8.0 and 8.6 million read pairs, as well as 6.7 and 7.5 million singleton reads were retained for the control and treatment libraries, respectively. 58.8–62.5 and 85.1–90.7 % of the reads were successfully mapped to the reference genome by TopHat and GSNAP. On average 19,652–26,008 transcripts encompassed at least one mapped fragment, with the mean number of mapped fragments 441.4–605.6 per transcript. Analysis of the obtained assemblies yielded 288 and 364 differentially expressed (DE) genes for TopHat and GSNAP respectively (see Additional files 1 and 2). The discrepancies between the two mapping methods could not be attributed to the potential differences in read assignment in cases of very similar paralogs as only one such unambiguous case was detected (data not shown). De novo reconstruction of full-length transcripts was limited due to moderate read coverage. Therefore, the contigs obtained by the reference-independent assembler Trinity were used only as a supplement to the two mapping-oriented approaches.
Lists of differentially expressed genes
Reference-independent and reference-guided TopHat and GSNAP assemblies yielded very similar lists of DE genes (data not shown), and all of the DE genes were previously annotated in the reference genome. For further analysis, the lists produced with these assembly strategies were merged producing two united lists for the two mapping programs (referred below as “TopHat” and “GSNAP” lists).
Overall, 435 genes were identified as differentially expressed by at least one approach. The two mapping approaches and the de novo assembly yielded similar lists of DE genes. 256 genes shown to be regulated by at least two approaches were considered the strongest candidates for differential expression (see Additional files 1 and 2). From the sets of DE genes we further excluded paralogous genes with nearly identical mRNA sequences, since the actual number of members being differentially expressed cannot be deduced for gene groups with high sequence similarities. A random representative of each group was retained.
The list of DE genes identified by at least two approaches is composed of a set of 230 genes: 158 up- and 72 down-regulated in the presence of the kairomone. Distribution of the l o g 2 fold changes in expression levels for these genes is shown in Fig. 2. Several genes were shown to be regulated in an on/off manner (i.e. showing no expression in either control or treatment) by individual assembly approaches. After averaging over the assemblies this strictly binary regulation was retained for only one of them: hxAUG26rep1s6g18t1, a protein without assigned function in the official gene set, for which a mollusc metallothionein family 2 signature was identified by InterProScan (see Additional file 1). Published results generated by qPCR (for predefined candidate genes) and tiled genomic microarrays to examine differentially expressed genes after treating Daphnia with Chaoborus kairomones [4, 65] were compared with our RNA-Seq dataset. All three lists show a low degree of overlap with the other two sets, but nevertheless the lists compiled from the results of the microarray and RNA-Seq analyses do share 31 genes with concordant patterns of expression, as well as four additional genes which show differential regulation in the opposite direction.

Frequency distribution of fold changes of DE genes. l o g 2 of the ratio “kairomone treatment”/“control” averaged over three assembly methods: GSNAP, TopHat and Trinity
Functional annotations
To functionally characterize DE genes, we used two independent sources of information: official gene annotations and InterProScan domain predictions.
In many cases even single amino-acid differences precluded domain identification in some otherwise identical proteins. Thus, to increase the power of the enrichment analyses, functional assignments and gene ontologies were interpolated from hits to individual members of paralogs to whole families of paralogous genes. As the gene family assignments provided with the official gene annotations were too broad, we performed independent, more stringent analysis of similarity between protein sequences.
Over-represented InterPro terms
Table 1 shows significantly over-represented domains and families as identified by InterProScan for the gene sets obtained with the aid of the two mapping methods. Among the up-regulated genes, genes coding for presumed cuticle-associated proteins (with 26–32 of them tagged as “insect cuticle proteins”) are most prevalent. Less abundant are proteins with evidence of lipid transport domains (lipoproteins), vitellogenin domains, and vWF domains, followed by genes coding for globins (together with cruorins) and cyclins.
The list of down-regulated candidates is enriched for genes coding for proteins with lectin-C, CUB, fibrinogen, collagen, TNF-like and complement C1q domains as well as proteins assigned to the GNS1/SUR4 family of unknown molecular function.
GO-enrichment analysis
Two sets of GO-term assignments were used in the GO-enrichment analysis: one deriving from the official gene annotations and another obtained with domain and family annotations reported by InterProScan. Corresponding lists of over-represented ontologies are shown in Tables 2 and 3. The terms obtained with InterProScan tend to be more precise and some of them have no corresponding categories in the second list, such as “regulation of cell cycle”, as well as the most abundant term pointing to cuticle-associated proteins. Over-representation of yolk proteins (“nutrient reservoir activity”) is explicitly detected as such only with the official annotations. For the down-regulated genes only one term was detected as being significantly over-represented with both annotation sources: carbohydrate binding, with collagen-related terms being additionally represented only in the InterPro-based list.
Chromatin and cell control proteins
Many of the up-regulated genes detected by RNA-Seq code for chromatin-associated or cell-cycle promoting proteins, although not all of the respective functions have been shown to be significantly over-represented (Fig. 3). Among them are nucleosome assembling proteins such as CAF-1, and histones H3 and H2b. Another histone H2b variant, distinct at the mRNA level (84.5 % identity), but similar on the amino-acid level (with the exception of the N-terminus, overall 77.2 % identity) is down-regulated. Cyclins encoded by three up-regulated genes belong to the A (1 gene) and B (2 genes) types.

Differentially expressed genes involved in cell-cycle control and chromatin regulation with their relative timing as known for other animals. The internal circle represents cell-cycle. The boxes corresponding to different proteins are arranged according to their role in progressing respective cell-cycle stages. The role of the F-box domain-containing protein can not be predicted with certainty. Numbers in parentheses represent numbers of differentially expressed genes belonging to respective families
Chemoreceptors and hormones
Among the DE genes detected by at least two assembly approaches only one is annotated as a gustatory receptor in the official gene set, hxAUG25p1s10g327t1 possessing a Scavenger receptor CD36 domain as reported in the wFleaBase and by InterProScan, with a fold change of 6.2 in our experiment. The only protein with identifiable humoral function in the RNA-Seq list of DE genes is an uncharacterized gene hxAUG26res18g88t1 with insulin-like domains identified by InterProScan. Its expression showed a 7.0 fold increase in the kairomone-treated juveniles (see Additional file 1).
Over-represented families of paralogs
In the list of genes shown to be differentially expressed by at least two approaches, 27 % of the up-regulated and 22 % of the down-regulated genes belong to families of paralogs. Some of these families are represented by several candidate genes in our list of DE genes and among them a considerable number of families is significantly over-represented (Table 4). The largest family among the up-regulated genes includes genes coding for products similar to a) pupal cuticle proteins, followed by b) Zinc-metalloproteinases, c) vitellogenin, d) a second family of cuticle-associated proteins, e) globin-cruorins, and f) other smaller families (Table 4). Products of gene families with significant down-regulation are characterized as a) C-type lectins, b) proteins of unknown molecular function with similarity to the C1q complement protein, c) proteins involved in elongation of very long chain fatty acids, and d) other less abundant families with nearly (Table 4).
Discussion
Gene expression patterns in the physiological context
Our RNA-Seq results generally agree with the hypothesis proposed in the introduction (see also Fig. 1):
-
1.
The most abundant and significantly over-represented functional group of up-regulated genes is composed of genes coding for cuticle-associated proteins (Tables 1, 2 and 4), which directly echoes morphological observations: i.e., production of neck-teeth (the main defence mechanism of the juvenile D. pulex), and changes in cuticle ultrastructure [26].
In addition, we observed increased transcription of genes involved in chromatin restructuring and the cell cycle (cyclins). This is likely related to the increased proliferative activity recently reported in the region underlying neck-teeth in induced D. pulex juveniles [66].
-
2.
A clear hint of resource re-allocation is suggested by the increase in expression of genes involved in lipid transport and metabolism, as well as globins. Six vitellogenin (precursor of the major yolk protein, vitellin [67, 68]) genes belong to this group as well. Production of yolk in daphnids starts as early as late juvenile stages, but the onset of vitellogenin mRNA synthesis takes place even earlier [69–71]. Vitellogenin is synthesized in fat bodies [72]; thus, the presence of residual maternal mRNA in our experiment can be excluded. In this respect, the increased expression of vitellogenin genes seems to point to one of the following factors or their combination: earlier onset of vitellogenesis, increased fecundity, or increased size of progeny. In a physiological study of vitellogenesis in D. magna, it was discovered that induction with the Chaoborus kairomone has no effect on the onset of yolk production, but decreases its rate [69]. Moreover, in a recent proteomics study of D. magna responses to another invertebrate predator, Triops, vitellogenin was shown to be among the proteins with decreased production in induced specimens [73]. These observations were based on protein content measurements for a distantly related species producing no neck-teeth, likely explaining theclear contradiction with our results. However,for D. pulex it was shown that kairomoneinduction leads to production of bigger offspring [29], which presumably requires a larger poolof yolk.
-
3.
Among functional groups significantly over-represented for the down-regulated genes, we find a large number of genes coding for proteins with domains that play various cellular roles: lectins, and proteins with CUB, fibrinogen and TNF domains. Intriguingly, proteins containing these domains function in immune responses in other invertebrates [74–76]. Although many details of molecular mechanisms of immune responses in Cladocera remain unknown [77, 78], decreased expressionof these proteins may be causally connected tothe observation that inducible defences inDaphnia lead to decreased resistance todiseases [79].
-
4.
Potential regulatory genes involved in metabolism of hormones and neurotransmitters or coding for chemoreceptors are nearly absent from our lists of differentially expressed genes. Among the up-regulated genes we found a gustatory receptor, which was designated as such in wFleaBase, but was not reported in an extensive in silico study of chemoreceptors in D. pulex [46] (see Additional file 1). We speculate that this receptor may be involved in perception of the kairomone, but this requires further experimental evidence.
No genes with identifiable roles in transcriptional regulation were discovered with our approach, which is likely related to the moderate sequencing coverage of the RNA-Seq.
Comparison to previous studies, future perspectives
A comparison of the lists of DE genes discovered in Chaoborus-induction experiments on juvenile D. pulex presented here with the results obtained from tiling microarrays shows that the two lists share a group of 31 genes showing concordant expression patterns. The overall discrepancy is nevertheless noticeable and may be attributed to differences in experimental set-up and/or water conditions and not to the differences in the platform, since comparative analyses generally reveal good correlation between microarray- and sequencing-based techniques [80–82]. Although both experiments were performed on the same D. pulex clone, the stage chosen for the microarray experiment was more advanced in comparison to the animals utilized in the current study. This observation signifies the necessity of an experiment involving several developmental stages to sample genes involved in different steps of the predator perception, signal perception and neck-teeth production and maintenance.
It is clear that any results obtained for a single genetic clone should not be over-extrapolated. More experiments on different D. pulex clones are necessary to make firm conclusions about the intraspecific variation of the genetic mechanisms acting in the anti-predatory response. Moreover, the existence of neck-teeth producing Daphnia species not directly related to D. pulex [21] calls for even broader sample of species to investigate the differences in trajectories ultimately leading to similar morphological features.
Conclusions
This study provides important insights into gene regulatory patterns underlying predator-induced defences, utilizing for the first time unbiased whole-transcriptome RNA-Seq expression data. In particular, our study characterizes different effector genes and gene families underlying morphological and life-history changes which are largely in agreement with expectations based on observed phenotypic changes. Our data represent the largest dataset on the genetic basis of anti-predator defences in Daphnia to date and add an important contribution to link a phenotypically plastic response directly with the underlying molecular genetic processes. A deeper understanding of these processes would be achieved with experiments on different genetic clones and at different developmental stages.
References
Schaack S. Daphnia comes of age: an ecological model in the genomic era. Mol Ecol. 2008; 17:1634–5.
Tollrian R, Leese F. Ecological genomics: steps towards unraveling the genetic basis of inducible defenses in Daphnia. BMC Biol. 2010; 8:51.
Altshuler I, Demiri B, Xu S, Constantin A, Yan ND, Cristescu ME. An integrated multi-disciplinary approach for studying multiple stressors in freshwater ecosystems: Daphnia as a model organism. Integr Comp Biol. 2011; 51:623–33.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, Bauer DJ, Cáceres CE, Carmel L, Casola C, Choi JH, Detter JC, Dong Q, Dusheyko S, Eads BD, Fröhlich T, Geiler-Samerotte KA, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva EV, Kültz D, Laforsch C, Lindquist E, Lopez J, Manak JR, Muller J, Pangilinan J, Patwardhan RP, Pitluck S, Pritham EJ, Rechtsteiner A, Rho M, Rogozin IB, Sakarya O, Salamov A, Schaack S, Shapiro H, Shiga Y, Skalitzky C, Smith Z, Souvorov A, Sung W, Tang Z, Tsuchiya D, Tu H, Vos H, Wang M, Wolf YI, Yamagata H, Yamada T, Ye Y, Shaw JR, Andrews J, Crease TJ, Tang H, Lucas SM, Robertson HM, Bork P, Koonin EV, Zdobnov EM, Grigoriev IV, Lynch M, Boore JL. The ecoresponsive genome of Daphnia pulex. Science. 2011; 331:555–61.
Lampert W. Daphnia: development of a model organism in ecology and evolution. Oldendorf/Luhe: International Ecology Institute; 2011.
Miner BE, De Meester L, Pfrender ME, Lampert W, Hairston NG. Linking genes to communities and ecosystems: Daphnia as an ecogenomic model. Proc Biol Sci. 2012; 279:1873–82.
Lampert W. Daphnia: model herbivore, predator and prey. Pol J Ecol. 2006; 54:607–20.
Seda J, Petrusek A. Daphnia as a model organism in limnology and aquatic biology: introductory remarks. J Limnol. 2011; 70:337–44.
Dodson SI. The ecological role of chemical stimuli for the zooplankton: predator-induced morphology in Daphnia. Oecologia. 1989; 78:361–7.
Tollrian R, Dodson SI. Inducible defenses in cladocera: constrains, costs, and multipredator environments In: Tollrian R, Harvell CD, editors. The ecology and evolution of inducible defenses. Princeton: Princeton University Press: 1999. p. 177–202.
Brönmark C, Pettersson LB, Nilsson PA. The ecology and evolution of inducible defenses In: Tollrian R, Harvell CD, editors. Princeton: Princeton University Press: 1999. p. 203–17.
Kuhlmann HW, Kusch J, Heckmann K. Predator-induced defenses in ciliated protozoa In: Tollrian R, Harvell CD, editors. The Ecology and Evolution of Inducible Defenses. Princeton: Princeton University Press: 1999. p. 142–59.
Tollrian R, Harvell CD. The evolution of inducible defenses: current ideas In: Tollrian R, Harvell CD, editors. The ecology and evolution of inducible defenses. Princeton: Princeton University Press: 1999. p. 306–21.
Stibor H, Lüning J. Predator-induced phenotypic variation in the pattern of growth and reproduction in Daphnia hyalina (Crustacea: Cladocera). Funct Ecol. 1994; 8:97–101.
Lüning J. Life-history responses to Chaoborus of spined and unspined Daphnia pulex. J Plankton Res. 1995; 17:71–84.
Boersma M, Spaak P, De Meester L. Predator-mediated plasticity in morphology, life history, and behavior of Daphnia: the uncoupling of responses. Am Nat. 1998; 152:237–48.
Weiss LC, Kruppert S, Laforsch C, Tollrian R. Chaoborus and Gasterosteus anti-predator responses in Daphnia pulex are mediated by independent cholinergic and gabaergic neuronal signals. PLoS ONE. 2012; 7:36879.
Beaton MJ, Hebert PDN. The cellular basis of divergent head morphologies in Daphnia. Limnol Oceanogr. 1997; 42:346–56.
Laforsch C, Tollrian R. Embryological aspects of inducible morphological defenses in Daphnia. J Morphol. 2004; 262:701–7.
Petrusek A, Tollrian R, Schwenk K, Haas A, Laforsch C. A “crown of thorns” is an inducible defense that protects Daphnia against an ancient predator. Proc Natl Acad Sci USA. 2009; 106:2248–52.
Juračka PJ, Laforsch C, Petrusek A. Neckteeth formation in two species of the Daphnia curvirostris complex (crustacea: Cladocera). J Limnol. 2011; 70:359–68.
Dodson SI. Adaptive change in plankton morphology in response to size-selective predation: a new hypothesis of cyclomorphosis. Limnol Oceanogr. 1974; 19:721–9.
Hebert PDN. The adaptive significance of cyclomorphosis in Daphnia: more possibilities. Freshw Biol. 1978; 8:313–20.
Hulsmann S, Vijverberg J, Boersma M, Mooij WM. Effects of infochemicals released by gape-limited fish on life history traits of Daphnia: a maladaptive response?J Plankton Res. 2004; 26:535–43.
Latta LC, Bakelar JW, Knapp RA, Pfrender ME. Rapid evolution in response to introduced predators II: the contribution of adaptive plasticity. BMC Evol Biol. 2007; 7:21.
Laforsch C, Ngwa W, Grill W, Tollrian R. An acoustic microscopy technique reveals hidden morphological defenses in Daphnia. Proc Natl Acad Sci USA. 2004; 101:15911–4.
Repka S, Walls M, Ketola M. Neck spine protects Daphnia pulex from predation by Chaoborus, but individuals with longer tail spine are at a greater risk. J Plankt Res. 1995; 17:393–403.
Tollrian R. Predator-induced morphological defenses: costs, life history shifts, and maternal effects in Daphnia pulex. Ecology. 1995; 76:1691–705.
Tollrian R. Chaoborus crystallinus predation on Daphnia pulex: can induced morphological changes balance effects of body size on vulnerability?Oecologia. 1995; 101:151–5.
Riessen HP. Chaoborus predation and delayed reproduction in Daphnia: a demographic modeling approach. Evol Ecol. 1999; 13:339–63.
Riessen HP. Costs of predator-induced morphological defences in Daphnia. Freshw Biol. 2012; 57:1422–33.
Dodson S. The ecological role of chemical stimuli for the zooplankton: predator-avoidance behavior in Daphnia. Limnol Oceanogr. 1988; 33:1431–9.
Boeing WJ, Ramcharan CW, Riessen HP. Multiple predator defence strategies in Daphnia pulex and their relation to native habitat. J Plankton Res. 2006; 28:571–84.
Dodson SI, Hanazato T, Gorski PR. Behavioral responses of Daphnia pulex exposed to carbaryl and Chaoborus kairomone. Environ Toxicol Chem. 1995; 14:43–50.
Dodson SI. Optimal swimming behavior of zooplankton In: Lenz P, Hartline DK, Purcell J, Macmillian D, editors. Zooplankton: Sensory Ecology and Physiology. Amsterdam: Gordon Breach Publishers: 1996. p. 365–74.
Weber A, van Noordwijk A. Swimming behaviour of Daphnia clones: differentiation through predator infochemicals. J Plankton Res. 2002; 24:1335–48.
Riessen HP, Sprules WG. Demographic costs of antipredator defenses in Daphnia pulex. Ecology. 1990; 71:1536–46.
Riessen HP. Cost-benefit model for the induction of an antipredator defense. Am Nat. 1992; 140:349–62.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009; 10:57–63.
Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011; 12:87–98.
McGettigan PA. Transcriptomics in the RNA-Seq era. Curr Opin Chem Biol. 2013; 17:4–11.
Pauwels K, Stoks R, De Meester L. Coping with predator stress: interclonal differences in induction of heat-shock proteins in the water flea Daphnia magna. J Evol Biol. 2005; 18:867–72.
Asselman J, De Coninck DI, Glaholt S, Colbourne J, Janssen C, Shaw J, De Schamphelaere K. Identification of pathways, gene networks and paralogous gene families in Daphnia pulex responding to exposure to the toxic cyanobacterium Microcystis aeruginosa. Environ Sci Technol. 2012; 46:8448–57.
Jansen M, Vergauwen L, Vandenbrouck T, Knapen D, Dom N, Spanier KI, Cielen A, De ML. Gene expression profiling of three different stressors in the water flea Daphnia magna. Ecotoxicology. 2013; 22(1):900–14.
De Coninck D, Asselman J, Glaholt S, Janssen C, Colbourne JK, Shaw J, De Schamphelaere K. Genome-wide transcription profiles reveal genotype-dependent responses of biological pathways and gene-families in Daphnia exposed to single and mixed stressors. Environ Sci Technol. 2014; 48:3513–522.
Peñalva-Arana DC, Lynch M, Robertson HM. The chemoreceptor genes of the waterflea Daphnia pulex: many grs but no ors. BMC Evol Biol. 2009; 9:79.
Thomson SA, Baldwin WS, Wang YH, Kwon G, Leblanc GA. Annotation, phylogenetics, and expression of the nuclear receptors in Daphnia pulex. BMC Genomics. 2009; 10:500.
Boucher P, Ditlecadet D, Dubé C, Dufresne F. Unusual duplication of the insulin-like receptor in the crustacean Daphnia pulex. BMC Evol Biol. 2010; 10:305.
Latta LC, Weider LJ, Colbourne JK, Pfrender ME. The evolution of salinity tolerance in Daphnia: a functional genomics approach. Ecol Lett. 2012; 15:794–802.
Colbourne JK, Crease TJ, Weider LJ, Hebert PDN, Dufresne F, Hobæk A. Phylogenetics and evolution of a circumarctic species complex (Cladocera: Daphnia pulex). Biol J Linn Soc. 1998; 65:347–65.
Vergilino R, Markova S, Ventura M, Manca M, Dufresne F. Reticulate evolution of the Daphnia pulex complex as revealed by nuclear markers. Mol Ecol. 2011; 20:1191–1207.
Trapnell C, Pachter L, Salzberg SL. Tophat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–11.
Wu TD, Nacu S. Fast and snp-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010; 26:873–81.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010; 28:511–5.
Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011; 27:2325–9.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011; 29:644–52.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012; 9:357–9.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucl Acids Res. 2005; 33:116–20.
Addou S, Rentzsch R, Lee D, Orengo CA. Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. J Mol Biol. 2009; 387:416–30.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-Seq: accounting for selection bias. Genome Biol. 2010; 11:14.
Kent WJ. Blat—the blast-like alignment tool. Genome Res. 2002; 12:656–64.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and samtools. Bioinformatics. 2009; 25:2078–9.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Gen Res. 2002; 12:996–1006.
Wilkinson L, Urbanek S. venneuler: Venn and Euler diagrams. R package version 1.1-0. 2011. http://CRAN.R-project.org/package=venneuler.
Miyakawa H, Imai M, Sugimoto N, Ishikawa Y, Ishikawa A, Ishigaki H, Okada Y, Miyazaki S, Koshikawa S, Cornette R, Miura T. Gene up-regulation in response to predator kairomones in the water flea, Daphnia pulex. BMC Dev Biol. 2010; 10:45.
Naraki Y, Hiruta C, Tochinai S. Identification of the precise kairomone-sensitive period and histological characterization of necktooth formation in predator-induced polyphenism in Daphnia pulex. Zoolog Sci. 2013; 30:619–25.
Zaffagnini F. Reproduction in Daphnia. Mem Inst Ital Idrobiol. 1987; 45:245–84.
Kato Y, Tokishita S, Ohta T, Yamagata H. A vitellogenin chain containing a superoxide dismutase-like domain is the major component of yolk proteins in cladoceran crustacean Daphnia magna. Gene. 2004; 334:157–65.
Stibor H. The role of yolk protein dynamics and predator kairomones for the life history of Daphnia magna. Ecology. 2002; 83:362–9.
Tokishita S, Kato Y, Kobayashi T, Nakamura S, Ohta T, Yamagata H. Organization and repression by juvenile hormone of a vitellogenin gene cluster in the crustacean, Daphnia magna. Biochem Biophys Res Commun. 2006; 345:362–70.
Kim J, Kim Y, Lee S, Kwak K, Chung WJ, Choi K. Determination of mRNA expression of DMRT93B, vitellogenin, and cuticle 12 in Daphnia magna and their biomarker potential for endocrine disruption. Ecotoxicology. 2011; 20:1741–8.
Zaffagnini F, Zeni C. Considerations on some cytological and ultrastructural observations on fat cells of Daphnia (Crustacea, Cladocera). Ital J Zool. 1986; 53:33–9.
Otte KA, Fröhlich T, Arnold GJ, Laforsch C. Proteomic analysis of Daphnia magna hints at molecular pathways involved in defensive plastic responses. BMC Genomics. 2014; 15:306.
Smith LC, Azumi K, Nonaka M. Complement systems in invertebrates. the ancient alternative and lectin pathways. Immunopharmacology. 1999; 42:107–20.
Fujita T, Matsushita M, Endo Y. The lectin-complement pathway – its role in innate immunity and evolution. Immunol Rev. 2004; 198:185–202.
Hanington PC, Zhang S. The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation. J Innate Immun. 2010; 3:17–27.
McTaggart SJ, Conlon C, Colbourne JK, Blaxter ML, Little TJ. The components of the Daphnia pulex immune system as revealed by complete genome sequencing. BMC Genomics. 2009; 10:175.
Auld SKJR. Physiology of the Cladocera In: Smirnov NN, editor. USA: Academic Press, Elsevier: 2014. p. 219–33.
Yin M, Laforsch C, Lohr JN, Wolinska J. Predator-induced defense makes Daphnia more vulnerable to parasites. Evolution. 2011; 65:1482–8.
Babbitt CC, Fedrigo O, Pfefferle AD, Boyle AP, Horvath JE, Furey TS, Wray GA. Both noncoding and protein-coding RNAs contribute to gene expression evolution in the primate brain. Genome Biol Evol. 2010; 2:67–79.
Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlén M, Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucl Acids Res. 2012; 40:10084–97.
Zhao S, Fung-Leung W, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE. 2014; 9:78644.
Acknowledgements
This work was supported in part by a grant of the Dinter-Foundation (Deutsches Stiftungszentrum) to FL and RT, startup funding of JRM and funding of RT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RT, LCW and FL designed the experiment; LCW performed kairomone induction; LCW and AR extracted the RNA; AR, MP and JRM performed the analysis. The manuscript was drafted by AR with significant input from FL, JRM and RT. All authors contributed to the discussion of the data and have read and approved the manuscript.
Additional files
Additional file 1
List of the differentially expressed genes. List of the 256 differentially expressed genes shown to be regulated in juvenile D. pulex as a response to the presence of Chaoborus larvae with at least two assembly methods. Gene names, locations and descriptions are according to the official gene annotations as provided by wFleaBase. InterPro term assignments are based on our analysis with the aid of InterProScan. The “+ref” suffixes refer to reference-guided versions of the respective mapping algorithms. Binary Excel File.
Additional file 2
Venn diagram of DE gene counts as identified by two mapping approaches: GSNAP and TopHat , and Trinity de novo assembly.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rozenberg, A., Parida, M., Leese, F. et al. Transcriptional profiling of predator-induced phenotypic plasticity in Daphnia pulex . Front Zool 12, 18 (2015). https://doi.org/10.1186/s12983-015-0109-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12983-015-0109-x